
CAMZYOS 2,5 mg, gélule, boîte de 2 plaquettes thermoformées de 14
Dernière révision : 30/05/2025
Taux de TVA : 2.1%
Prix de vente : 1 383,69 €
Taux remboursement SS : 65%
Base remboursement SS : 1 383,69 €
Laboratoire exploitant : BRISTOL-MYERS SQUIBB
Source :

CAMZYOS est indiqué chez les patients adultes pour le traitement de la cardiomyopathie hypertrophique obstructive (CMHo) symptomatique (stade II-III de la classification NYHA, New York Heart Association) (voir rubrique Propriétés pharmacodynamiques).
- Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception efficace (voir rubriques Mises en garde spéciales et précautions d'emploi et Fertilité, grossesse et allaitement).
- Traitement concomitant par des inhibiteurs puissants du CYP3A4 chez les patients présentant un phénotype métaboliseur lent du CYP2C19 ou un phénotype non déterminé du CYP2C19 (voir rubriques Posologie et mode d'administration, Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
-
Traitement concomitant par un inhibiteur puissant du CYP2C19 en association avec un inhibiteur puissant du CYP3A4 (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Dysfonctionnement systolique défini par une FEVG symptomatique < 50 %
Le mavacamten réduit la FEVG et peut provoquer une insuffisance cardiaque due à un dysfonctionnement systolique, définie par une FEVG symptomatique < 50 %. Les patients présentant une affection intercurrente grave telle qu'une infection ou une arythmie (y compris une fibrillation atriale ou une autre tachyarythmie non contrôlée) ou faisant l'objet d'une chirurgie cardiaque majeure sont susceptibles d'être exposés à un risque accru de dysfonctionnement systolique et d'évoluer vers une insuffisance cardiaque (voir rubrique Effets indésirables). Une dyspnée nouvelle ou aggravée, des douleurs thoraciques, de la fatigue, des palpitations, un œdème des jambes ou des élévations du peptide natriurétique de type N-terminal-pro-B (NT-proBNP) peuvent être des signes et symptômes d'un dysfonctionnement systolique et doivent inciter à mener une évaluation de la fonction cardiaque. La FEVG doit être mesurée avant l'instauration du traitement et être étroitement surveillée par la suite. L'interruption du traitement peut être nécessaire pour s'assurer que la FEVG reste ≥ 50 % (voir rubrique Posologie et mode d'administration).
Risque d'insuffisance cardiaque ou perte de réponse thérapeutique au mavacamten en raison d'interactions
Le mavacamten est principalement métabolisé par le CYP2C19 et dans une moindre mesure par le CYP3A4, et principalement par le CYP3A4 chez les métaboliseurs lents du CYP2C19, ce qui peut donner lieu aux interactions suivantes (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) :
- L'instauration ou l'augmentation de dose d'un inhibiteur puissant ou modéré du CYP3A4 ou de tout inhibiteur du CYP2C19 peut augmenter le risque d'insuffisance cardiaque due à un dysfonctionnement systolique.
- L'arrêt ou la diminution de la dose de tout inhibiteur du CYP3A4 ou du CYP2C19 peut entraîner une perte de réponse thérapeutique au mavacamten.
- L'instauration d'un inducteur puissant du CYP3A4 ou d'un inducteur puissant du CYP2C19 peut entraîner une perte de réponse thérapeutique au mavacamten.
- L'arrêt d'un inducteur puissant du CYP3A4 ou d'un inducteur puissant du CYP2C19 peut augmenter le risque d'insuffisance cardiaque due à un dysfonctionnement systolique.
Avant et
pendant le traitement par mavacamten, le risque d'interactions doit être pris
en compte, y compris avec les médicaments en vente libre (tels que l'oméprazole
ou l'ésoméprazole).
- Un traitement concomitant par des inhibiteurs puissants du CYP3A4 chez les patients présentant un phénotype métaboliseur lent du CYP2C19 et un phénotype non déterminé du CYP2C19 est contre-indiqué (voir rubrique Contre-indications).
- Un traitement concomitant par un inhibiteur puissant du CYP2C19 en association avec un inhibiteur puissant du CYP3A4 est contre-indiqué (voir rubrique Contre-indications).
- Un ajustement de la dose de mavacamten et/ou une surveillance étroite peuvent être nécessaires en cas d'instauration, d'arrêt ou de modification de dose d'un traitement concomitant avec des inhibiteurs ou des inducteurs du CYP2C19 ou du CYP3A4 (voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions). L'administration intermittente de ces médicaments n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Utilisation concomitante d'inotropes négatifs
La sécurité de l'utilisation concomitante du mavacamten avec le disopyramide, ou de l'utilisation du mavacamten chez les patients traités par des bêta-bloquants en association avec le vérapamil ou le diltiazem, n'a pas été établie. Il est donc recommandé de réaliser une surveillance étroite des patients en cas de traitement concomitant par ces médicaments (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Toxicité embryo-fœtale
D'après les
études menées sur les animaux, le mavacamten est susceptible de provoquer une
toxicité embryo-fœtale lorsqu'il est administré chez la femme enceinte (voir
rubrique Données de sécurité préclinique). En raison des risques pour le
fœtus, CAMZYOS est contre-indiqué pendant la grossesse et chez les femmes en
âge de procréer n'utilisant pas de contraception efficace. Avant l'instauration
du traitement, les femmes en âge de procréer doivent être informées des risques
pour le fœtus, doivent obtenir un résultat négatif à un test de grossesse et
doivent utiliser une contraception efficace pendant le traitement et pendant 6
mois après l'arrêt du traitement (voir rubriques Contre-indications et Fertilité,
grossesse et allaitement).
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés avec le mavacamten sont les étourdissements (17 %), la dyspnée (12 %), le dysfonctionnement systolique (5 %) et la syncope (5 %).
Tableau des effets indésirables
Le tableau ci-dessous présente les effets indésirables survenant chez les patients traités par mavacamten dans deux études de phase III (EXPLORER-HCM et VALOR-HCM). Au total, 179 patients ont reçu une dose journalière de 2,5 mg, 5 mg, 10 mg ou 15 mg de mavacamten. La durée médiane de traitement pour les patients recevant le mavacamten a été de 30,1 semaines (durée : de 1,6 à 40,3 semaines).
Les effets indésirables inclus dans le tableau 3 sont répertoriés par classe de systèmes d'organes selon la base de données MedDRA. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont présentés par ordre décroissant de fréquence et de gravité. En outre, la catégorie correspondante en matière de fréquence pour chaque effet indésirable est définie comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000).
Tableau 3 : Effets indésirables
| Classe de systèmes d'organes | Effet indésirable | Fréquence |
| Affections du système nerveux | Etourdissement | Très fréquent |
| Syncope | Fréquent | |
| Affections cardiaques | Dysfonctionnement systoliquea | Fréquent |
| Affections respiratoires, thoraciques et médiastinales | Dyspnée | Très fréquent |
a Défini par une FEVG < 50 % avec ou sans symptômes.
Description des effets indésirables sélectionnés
Dysfonctionnement systolique
Dans
les études cliniques de phase III, 5 % (9/179) des patients du groupe
mavacamten ont enregistré des diminutions réversibles de la FEVG <
50 % (médiane 45 % : de 35 à 49 %) pendant le traitement. Chez 56 %
(5/9) de ces patients, les diminutions ont été observées sans autres
manifestations cliniques. La FEVG a été rétablie chez tous les patients
traités par mavacamten après l'interruption du mavacamten, et ils sont
parvenus au terme de l'étude en poursuivant le traitement (voir
rubrique Mises en garde spéciales et précautions d'emploi).
Dyspnée
Dans les études cliniques de phase III, une dyspnée a été rapportée chez 12,3 % des patients traités par mavacamten versus 8,7
% des patients sous placebo. Dans l'étude EXPLORER-HCM, la plupart des
événements de dyspnée (67 %) ont été rapportés après l'arrêt du
mavacamten, avec un délai médian d'apparition de 2 semaines (de 0,1 à
4,9) après la dernière dose.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT L'INSTAURATION DU TRAITEMENT :
- Evaluer la fraction d'éjection ventriculaire gauche (FEVG) par échocardiographie. Si la FEVG est < 55 %, le traitement ne doit pas être instauré.
- Obtenir un résultat de test de grossesse négatif chez les femmes en âge de procréer. Les INFORMER des risques graves pour le fœtus.
- Déterminer le phénotype du cytochrome P450 2C19 (CYP2C19) des patients par génotypage afin d'identifier la dose de mavacamten appropriée. Les patients présentant un phénotype métaboliseur lent du CYP2C19 peuvent être confrontés à une exposition accrue au mavacamten (jusqu'à 3 fois plus), ce qui peut augmenter le risque de dysfonctionnement systolique comparé aux métaboliseurs normaux. En cas d'instauration du traitement avant la détermination du phénotype du CYP2C19, les patients doivent suivre les instructions posologiques des métaboliseurs lents jusqu'à la détermination du phénotype du CYP2C19.
SURVEILLANCE du traitement :
- Evaluer la réponse clinique précoce chez le patient par le gradient de la chambre de chasse ventriculaire gauche (CCVG), avec manœuvre de Valsalva, 4 et 8 semaines après l'instauration du traitement. Une fois la dose de maintenance individualisée atteinte, avec une FEVG ≥ 55 %, les patients doivent faire l'objet d'une évaluation tous les 6 mois. Pour les patients présentant une FEVG comprise entre 50 et < 55 %, quel que soit le gradient CCVG avec manoeuvre de Valsalva, une évaluation doit être effectuée tous les 3 mois. Si, lors d'une visite, le patient présente une FEVG < 50 %, le traitement devra être interrompu pendant 4 semaines et jusqu'à ce que la FEVG revienne à une valeur ≥ 50 %.
- Confirmer par échographie que la FEVG est ≥ 50 %.
REMETTRE le guide patients contenant la carte patients.
Risque d'insuffisance cardiaque ou perte de réponse thérapeutique au mavacamten en raison d'INTERACTIONS :
Le mavacamten est principalement métabolisé par le CYP2C19 et dans une moindre mesure par le CYP3A4, et principalement par le CYP3A4 chez les métaboliseurs lents du CYP2C19.
Ainsi, avant et pendant le traitement par mavacamten, le risque d'interactions doit être pris en compte.
- Evaluer la fraction d'éjection ventriculaire gauche (FEVG) par échocardiographie. Si la FEVG est < 55 %, le traitement ne doit pas être instauré.
- Obtenir un résultat de test de grossesse négatif chez les femmes en âge de procréer. Les INFORMER des risques graves pour le fœtus.
- Déterminer le phénotype du cytochrome P450 2C19 (CYP2C19) des patients par génotypage afin d'identifier la dose de mavacamten appropriée. Les patients présentant un phénotype métaboliseur lent du CYP2C19 peuvent être confrontés à une exposition accrue au mavacamten (jusqu'à 3 fois plus), ce qui peut augmenter le risque de dysfonctionnement systolique comparé aux métaboliseurs normaux. En cas d'instauration du traitement avant la détermination du phénotype du CYP2C19, les patients doivent suivre les instructions posologiques des métaboliseurs lents jusqu'à la détermination du phénotype du CYP2C19.
SURVEILLANCE du traitement :
- Evaluer la réponse clinique précoce chez le patient par le gradient de la chambre de chasse ventriculaire gauche (CCVG), avec manœuvre de Valsalva, 4 et 8 semaines après l'instauration du traitement. Une fois la dose de maintenance individualisée atteinte, avec une FEVG ≥ 55 %, les patients doivent faire l'objet d'une évaluation tous les 6 mois. Pour les patients présentant une FEVG comprise entre 50 et < 55 %, quel que soit le gradient CCVG avec manoeuvre de Valsalva, une évaluation doit être effectuée tous les 3 mois. Si, lors d'une visite, le patient présente une FEVG < 50 %, le traitement devra être interrompu pendant 4 semaines et jusqu'à ce que la FEVG revienne à une valeur ≥ 50 %.
- Confirmer par échographie que la FEVG est ≥ 50 %.
REMETTRE le guide patients contenant la carte patients.
Risque d'insuffisance cardiaque ou perte de réponse thérapeutique au mavacamten en raison d'INTERACTIONS :
Le mavacamten est principalement métabolisé par le CYP2C19 et dans une moindre mesure par le CYP3A4, et principalement par le CYP3A4 chez les métaboliseurs lents du CYP2C19.
Ainsi, avant et pendant le traitement par mavacamten, le risque d'interactions doit être pris en compte.
INFORMER IMMÉDIATEMENT LE MÉDECIN ou
LE PHARMACIEN en cas de :
- essoufflement nouveau ou aggravé,
- douleur thoracique,
- fatigue,
- palpitations (battements cardiaques forts qui peuvent être rapides ou
irréguliers),
- gonflement des jambes,
- infection grave,
- battements cardiaques irréguliers.
PRUDENCE en cas de consommation de jus de pamplemousse ou de préparation à base
de plantes contenant du millepertuis (Hypericum
perforatum).
NE PAS CONDUIRE de véhicules ou UTILISER de machines en cas d'étourdissement.
PATIENTE EN AGE D'AVOIR DES ENFANTS : UTILISER
une contraception efficace pendant le traitement et pendant les 6 mois qui
suivent l'arrêt de celui-ci.
Femmes en âge de procréer / Contraception chez les femmes
CAMZYOS est contre-indiqué chez les femmes en âge de procréer n'utilisant pas de contraception efficace (voir rubrique Contre-indications). Par conséquent, avant l'instauration du traitement chez les femmes en âge de procréer, le résultat du test de grossesse doit être négatif et la patiente doit être informée des risques graves pour le fœtus. Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et pendant 6 mois après l'arrêt de CAMZYOS, car le délai d'élimination du mavacamten dans l'organisme est d'environ 5 demi-vies (environ 45 jours pour les métaboliseurs normaux du CYP2C19 et 115 jours pour les métaboliseurs lents du CYP2C19) après l'arrêt du traitement (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
L'éventualité d'un retour de l'obstruction de la CCVG et de l'importance des symptômes doit être considérée lors de l'arrêt du traitement par mavacamten dans le cadre de la planification d'une grossesse (voir rubrique Mises en garde spéciales et précautions d'emploi).
Grossesse
Il n'existe pas de données sur l'utilisation du mavacamten chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Le mavacamten est susceptible de provoquer une toxicité embryo-fœtale lorsqu'il est administré pendant la grossesse. Par conséquent, CAMZYOS est contre-indiqué pendant la grossesse (voir rubrique Contre-indications). CAMZYOS doit être arrêté 6 mois avant le début de grossesse envisagé (voir rubrique Mises en garde spéciales et précautions d'emploi). Si une patiente tombe enceinte, le mavacamten doit être arrêté. Des conseils médicaux doivent être donnés concernant le risque d'effets délétères pour le fœtus associé au traitement et des examens d'échographie devront être réalisés.
Allaitement
On ne sait pas si le mavacamten ou ses métabolites sont excrétés dans le lait maternel. Il n'existe pas de données sur l'excrétion du mavacamten ou de ses métabolites dans le lait animal (voirrubrique Données de sécurité préclinique). Les conséquences en termes d'effets indésirables sur les nouveau-nés/nourrissons allaités étant inconnues , les femmes ne doivent pas allaiter pendant le traitement par mavacamten.
Fertilité
Aucune donnée sur la fertilité humaine avec le mavacamten n'est disponible. Les études effectuées chez l'animal sont insuffisantes en ce qui concerne la fertilité chez les mâles ou les femelles (voir rubrique Données de sécurité préclinique).
Interactions pharmacodynamiques
Si un traitement par un nouvel inotrope négatif est instauré, ou si la dose d'un inotrope négatif est augmentée chez un patient recevant le mavacamten, une surveillance médicale étroite avec un contrôle de la FEVG doit être assurée jusqu'à ce que des doses stables et une réponse clinique soient obtenues (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Interactions pharmacocinétiques
Effet d'autres médicaments sur le mavacamten
Chez les métaboliseurs intermédiaires, normaux,
rapides et ultrarapides du CYP2C19, le mavacamten est
principalement métabolisé par le CYP2C19 et dans une moindre mesure par le
CYP3A4. Chez les métaboliseurs lents du CYP2C19, le
métabolisme se fait principalement par le CYP3A4 (voir rubrique Propriétés
pharmacocinétiques). Les inhibiteurs/inducteurs du CYP2C19 et les
inhibiteurs/inducteurs du CYP3A4 sont donc susceptibles d'affecter la clairance
du mavacamten et d'augmenter/diminuer la
concentration plasmatique du mavacamten en fonction
du phénotype du CYP2C19.
Toutes les études cliniques d'interactions médicamenteuses ont été menées
principalement chez des métaboliseurs normaux du
CYP2C19, sans inclure aucun métaboliseur lent du
CYP2C19 dans l'évaluation. Par conséquent, l'effet de la co-administration
d'inhibiteurs du CYP2C19 et du CYP3A4 avec le mavacamten
chez les métaboliseurs lents du CYP2C19 n'est pas
parfaitement connu.
Les recommandations relatives à la modification de la dose et/ou à une
surveillance supplémentaire en cas d'instauration, d'arrêt ou de modification
de dose d'un traitement concomitant avec des inhibiteurs du CYP2C19 ou du
CYP3A4, ou des inducteurs du CYP2C19 ou du CYP3A4, sont indiquées dans le
tableau 2.
Association d'inhibiteurs puissants du CYP2C19 avec des
inhibiteurs puissants du CYP3A4
La co-administration du mavacamten
avec un inhibiteur puissant du CYP2C19 associé à un inhibiteur puissant du
CYP3A4 est contre-indiquée (voir rubrique Contre-indications).
Inhibiteurs du CYP2C19
L'effet d'un inhibiteur modéré et puissant du CYP2C19 sur la pharmacocinétique
du mavacamten n'a pas été évalué dans le cadre d'une
étude clinique d'interactions médicamenteuses. L'effet d'un inhibiteur puissant
du CYP2C19 (par ex. la ticlopidine) sera comparable à
celui observé avec un statut de métaboliseur lent du
CYP2C19 (voir tableau 1).
La co-administration du mavacamten
avec un inhibiteur faible du CYP2C19 (oméprazole) a
entraîné une augmentation de 48 % de l'ASCinf du mavacamten sans effet sur la Cmax
chez les métaboliseurs normaux du CYP2C19.
L'administration intermittente d'un inhibiteur du CYP2C19 (tel que l'oméprazole ou l'ésoméprazole)
n'est pas recommandée (voir rubrique Mises en garde spéciales et précautions
d'emploi).
Inhibiteurs du CYP3A4
L’effet
d’inhibiteurs puissants du CYP3A4 sur la pharmacocinétique du
mavacamten n’a pas été évalué dans le cadre d’une étude clinique
d’interactions médicamenteuses. La co-administration du mavacamten avec
un inhibiteur puissant du CYP3A4 (itraconazole) chez les métaboliseurs
normaux du CYP2C19 devrait entraîner une augmentation de la
concentration plasmatique du mavacamten allant jusqu’à 59 % et 40 % de
l’ASC0-24 et de la Cmax, respectivement.
La co-administration du mavacamten et d’un inhibiteur modéré du CYP3A4
(vérapamil) chez les métaboliseurs normaux du CYP2C19 a entraîné une
augmentation de la concentration plasmatique du mavacamten de 16 % et
52 % de l’ASCinf et de la Cmax, respectivement. Cette variation n’a pas
été considérée comme cliniquement significative.
Inducteurs du CYP2C19 et CYP3A4
Aucune étude clinique d'interaction n'a été menée pour examiner l'effet d'une
administration concomitante avec un inducteur puissant du CYP3A4 et du CYP2C19.
La co-administration du mavacamten
avec un inducteur puissant du CYP2C19 et du CYP3A4 (par ex. la rifampicine)
devrait affecter significativement la pharmacocinétique (PK) du mavacamten et conduit à une diminution de l'efficacité ;
par conséquent, la co-administration avec des
inducteurs puissants du CYP2C19 et du CYP3A4 n'est pas recommandée. En cas
d'arrêt du traitement concomitant avec un inducteur puissant du CYP2C19 ou du
CYP3A4, augmenter le nombre d'évaluations cliniques et diminuer la dose de mavacamten (voir rubrique Posologie et mode
d'administration).
Tableau 2 : Modification de la posologie/surveillance du mavacamten en fonction des
traitements concomitants
|
Traitement
concomitant |
Phénotype métaboliseur lent du CYP2C19* |
Phénotype
métaboliseur intermédiaire, normal, rapide et ultrarapide du CYP2C19 |
| Inhibiteurs | ||
|
Traitement
concomitant avec un inhibiteur puissant du CYP2C19 et un inhibiteur puissant du CYP3A4 |
Contre-indiqué (voir rubrique Contre-indications) |
Contre-indiqué
(voir rubrique Contre-indications) |
|
Inhibiteur
puissant du CYP2C19 (par ex. ticlopidine, fluconazole, fluvoxamine) |
Aucun
ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). Si le phénotype du CYP2C19 n'a pas encore été déterminé : Aucun ajustement de la dose initiale de 2,5 mg n'est nécessaire. Réduire la dose de 5 mg à 2,5 mg, ou interrompre le traitement s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
Instaurer
le mavacamten à une dose de 2,5 mg. Réduire la dose de 15 mg à 5 mg, de 10 mg à 2,5 mg et de 5 mg à 2,5 mg, ou interrompre le traitement s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
|
Inhibiteur
puissant du CYP3A4 (par ex. clarithromycine, itraconazole, kétoconazole, voriconazole, ritonavir, cobicistat, céritinib, idélalisib, tucatinib) |
Contre-indiqué (voir rubrique Contre-indications) |
Aucun
ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
|
Inhibiteur
modéré du CYP2C19 (par ex. fluconazole, fluoxétine, oméprazolea) |
Aucun
ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). Si le phénotype du CYP2C19 n'a pas encore été déterminé : Aucun ajustement de la dose initiale de 2,5 mg n'est nécessaire.Réduire la dose de 5 mg à 2,5 mg ou interrompre le traitement s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). |
Aucun
ajustement de la dose initiale de 5 mg n'est nécessaire. Instauration ou augmentation de dose d'un inhibiteur modéré pendant le traitement par mavacamten : Réduire la dose d'un niveau de dose ou interrompre le traitement s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
|
Inhibiteur
modéré du CYP3A4 (par ex. érythromycine, jus de pamplemousse, vérapamil, diltiazem) |
Si
un traitement est en cours lors de l'instauration du traitement par mavacamten, aucun ajustement de la dose initiale de 2,5 mg n'est nécessaire. Instauration ou augmentation de dose d'un inhibiteur modéré pendant le traitement par mavacamten : Si le patient reçoit une dose de 5 mg de mavacamten, réduire sa dose à 2,5 mg ou interrompre le traitement pendant 4 semaines s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
Aucun
ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
|
Inhibiteur
faible du CYP2C19 (par ex. cimétidine, citalopram, oméprazolea, ésoméprazole) |
Aucun
ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). |
Instauration
ou augmentation de dose d'un inhibiteur faible pendant le traitement par mavacamten : Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). |
|
Inhibiteur
faible du CYP3A4 (par ex. cimétidine, ésoméprazole, oméprazole, pantoprazole) |
Si
un traitement est en cours lors de l'instauration du traitement par mavacamten, aucun ajustement de la dose initiale de 2,5 mg n'est nécessaire. Instauration ou augmentation de dose d'un inhibiteur faible pendant le traitement par mavacamten : Si le patient reçoit une dose de 5 mg de mavacamten, réduire sa dose à 2,5 mg ou interrompre le traitement pendant 4 semaines s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
Instauration
ou augmentation de dose d'un inhibiteur faible pendant le traitement par mavacamten : Aucun ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). |
| Inducteurs | ||
|
Inducteur
puissant du CYP2C19 et inducteur puissant du CYP3A4 (par ex. rifampicine, apalutamide, enzalutamide, mitotane, phénytoïne, carbamazépine, éfavirenz, millepertuis) |
Instauration
ou augmentation de dose d'un inducteur puissant pendant le traitement par mavacamten : Surveiller le gradient CCVG et la FEVG 4 semaines plus tard. Ajuster la dose de mavacamten en fonction de l'évaluation clinique, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). La dose maximale est de 5 mg. Interruption ou diminution de dose d'un inducteur puissant pendant le traitement par mavacamten : Réduire la dose de mavacamten de 5 mg à 2,5 mg ou interrompre le traitement s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
Instauration
ou augmentation de dose d'un inducteur puissant pendant le traitement par mavacamten : Surveiller le gradient CCVG et la FEVG 4 semaines plus tard. Ajuster la dose de mavacamten en fonction de l'évaluation clinique, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). Interruption ou diminution de dose d'un inducteur puissant pendant le traitement par mavacamten : Réduire le mavacamten d'un niveau de dose lorsque le traitement est à 5 mg ou plus. Maintenir la dose de mavacamten lorsqu'elle est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
|
Inducteur
modéré ou faible du CYP2C19 (par ex. letermovir, noréthistérone, prednisone) |
Aucun
ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). |
Instauration
de dose d'un inducteur modéré ou faible pendant le traitement par mavacamten : Surveiller le gradient CCVG et la FEVG 4 semaines plus tard. Ajuster la dose de mavacamten en fonction de l'évaluation clinique, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). Interruption d'un inducteur modéré ou faible pendant le traitement par mavacamten : Réduire le mavacamten d'un niveau de dose lorsque le traitement est à 5 mg ou plus. Maintenir la dose de mavacamten lorsqu'elle est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). |
|
Inducteur
modéré ou faible du CYP3A4 (par * ex. phénobarbital, primidone) |
Instauration
ou augmentation de dose d'un inducteur modéré ou faible pendant le traitement par mavacamten : Surveiller le gradient CCVG et la FEVG 4 semaines plus tard. Ajuster la dose de mavacamten en fonction de l'évaluation clinique, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). Interruption ou diminution de dose d'un inducteur modéré ou faible pendant le traitement par mavacamten : Réduire la dose de mavacamten à 2,5 mg ou interrompre le traitement s'il est à 2,5 mg. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient (voir rubrique Posologie et mode d'administration). |
Aucun
ajustement posologique. Surveiller la FEVG 4 semaines plus tard, puis reprendre le calendrier de suivi et de titration du patient. Ajuster la dose de mavacamten en fonction de l'évaluation clinique (voir rubrique Posologie et mode d'administration). |
* inclut les patients dont le phénotype du CYP2C19 n'a pas encore été déterminé.
a l'oméprazole est considéré comme un inhibiteur faible du CYP2C19 à une dose de 20 mg une fois par jour et comme un inhibiteur modéré du CYP2C19 à une dose totale journalière de 40 mg.
Effet du mavacamten sur d'autres
médicaments
Les données in vitro du mavacamten indiquent
une induction potentielle du CYP3A4. La
co-administration d'une cure de 17 jours de mavacamten, à des expositions cliniquement pertinentes,
chez des métaboliseurs normaux, rapides et
ultrarapides du CYP2C19, n'a pas diminué l'exposition à l'éthinylœstradiol
et à la noréthistérone, qui sont les composants de
contraceptifs oraux courants et les substrats du CYP3A4. En outre, la co-administration d'une cure de 16 jours de mavacamten chez des métaboliseurs
normaux du CYP2C19, à des expositions cliniquement pertinentes, a entraîné une
diminution de 13 % de la concentration plasmatique de midazolam.
Cette variation n'a pas été considérée comme cliniquement significative.
Le traitement doit être instauré sous la surveillance d'un médecin expérimenté dans la prise en charge de patients atteints de cardiomyopathie.
Avant l'instauration du traitement, la fraction d'éjection ventriculaire gauche (FEVG) du patient doit faire l'objet d'une évaluation par échocardiographie (voir rubrique Mises en garde spéciales et précautions d'emploi). Si la FEVG est < 55 %, le traitement ne doit pas être instauré.
Avant l'instauration du traitement, les femmes en âge de procréer doivent présenter un test de grossesse négatif (voir rubriques Mises en garde spéciales et précautions d'emploi et Fertilité, grossesse et allaitement).
Le phénotype du cytochrome P450 2C19 (CYP2C19) des patients devrait être déterminé par génotypage afin d'identifier la dose de mavacamten appropriée. Les patients présentant un phénotype métaboliseur lent du CYP2C19 peuvent être confrontés à une exposition accrue au mavacamten (jusqu'à 3 fois plus), ce qui peut augmenter le risque de dysfonctionnement systolique comparé aux métaboliseurs normaux (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques). En cas d'instauration du traitement avant la détermination du phénotype du CYP2C19, les patients doivent suivre les instructions posologiques des métaboliseurs lents (voir figure 1 et 3 et tableau 1) jusqu'à la détermination du phénotype du CYP2C19.
Posologie
L'intervalle de dose est compris entre 2,5 mg et 15 mg (soit 2,5 mg, 5 mg, 10 mg ou 15 mg).
Phénotype métaboliseur lent du CYP2C19
La posologie initiale recommandée est de 2,5 mg par voie orale une fois par jour. La dose maximale est de 5 mg une fois par jour. Il convient d'évaluer la réponse clinique précoce chez le patient par le gradient de la chambre de chasse ventriculaire gauche (CCVG), avec manœuvre de Valsalva, 4 et 8 semaines après l'instauration du traitement (voir figure 1).
Phénotypes métaboliseurs intermédiaires, normaux, rapides et ultrarapides du CYP2C19
La posologie initiale recommandée est de 5 mg par voie orale une fois par jour. La dose maximale est de 15 mg une fois par jour. Il convient d'évaluer la réponse clinique précoce chez le patient par le gradient CCVG avec manœuvre de Valsalva, 4 et 8 semaines après l'instauration du traitement (voir figure 2).
Une fois la dose de maintenance individualisée atteinte avec une FEVG ≥ 55 %, les patients doivent faire l'objet d'une évaluation tous les 6 mois. Pour les patients présentant une FEVG comprise entre 50 et < 55 %, quel que soit le gradient CCVG avec manœuvre de Valsalva, une évaluation doit être effectuée tous les 3 mois (voir figure 3). Si, lors d'une visite, le patient présente une FEVG < 50 %, le traitement devra être interrompu pendant 4 semaines et jusqu'à ce que la FEVG revienne à une valeur ≥ 50 % (voir figure 4).
Chez les patients présentant une affection intercurrente telle qu'une infection grave ou une arythmie (y compris une fibrillation atriale ou une autre tachyarythmie non contrôlée) susceptible d'altérer la fonction systolique, il est recommandé d'effectuer une évaluation de la FEVG ; par ailleurs, les augmentations de dose ne sont pas recommandées tant que l'affection intercurrente n'est pas résolue (voir rubrique Mises en garde spéciales et précautions d'emploi).
Il faut envisager d'arrêter le traitement chez les patients qui n'ont montré aucune réponse (par ex. aucune amélioration des symptômes, de la qualité de vie, de la capacité à faire de l'exercice physique ou du gradient CCVG) après 4-6 mois à la dose maximale tolérée.
Figure 1 : Instauration du
traitement pour le phénotype métaboliseur lent du CYP2C19
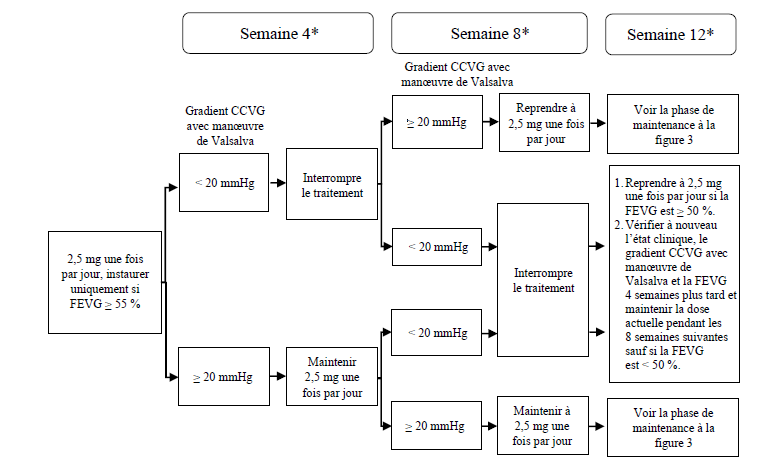
* Interrompre le traitement si, lors d'une visite clinique, la FEVG est < 50 % ; reprendre le traitement après 4 semaines si la FEVG est ≥ 50 % (voir figure 4).
CCVG = chambre de chasse ventriculaire gauche ; FEVG = fraction d'éjection ventriculaire gauche
Figure 2 : Instauration du traitement pour le phénotype métaboliseur intermédiaire, normal, rapide et ultrarapide du CYP2C19
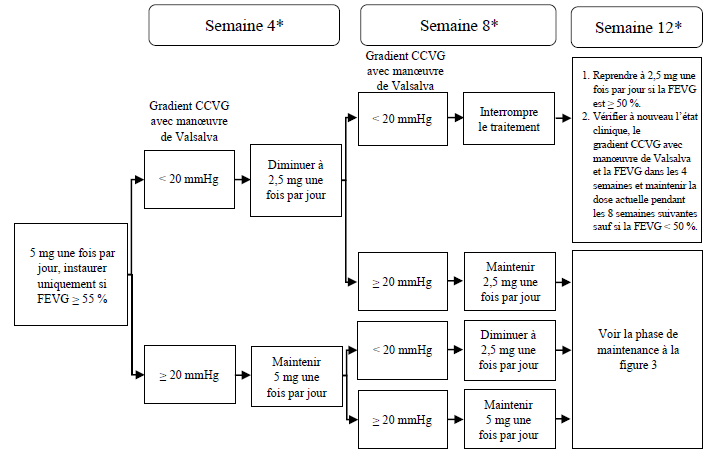
* Interrompre le traitement si, lors d'une visite clinique, la FEVG est < 50 % ; reprendre le traitement après 4 semaines si la FEVG est ≥ 50 % (voir figure 4).
CCVG = chambre de chasse ventriculaire gauche ; FEVG = fraction d'éjection ventriculaire gauche
Figure 3 : Phase de maintenance
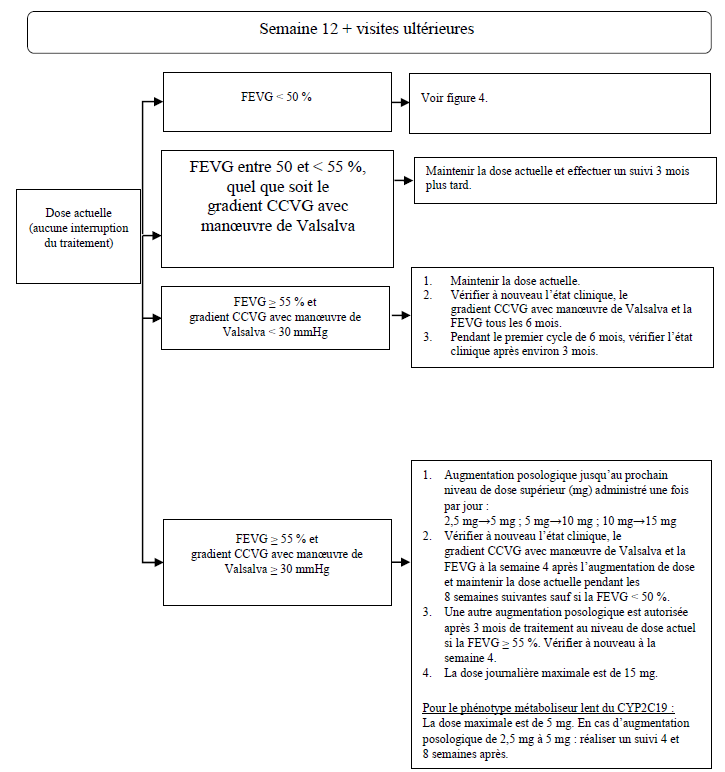
CCVG = chambre de chasse ventriculaire gauche ; FEVG = fraction d'éjection ventriculaire gauche
Figure 4 : Interruption du traitement lors d'une visite clinique si la FEVG < 50 %
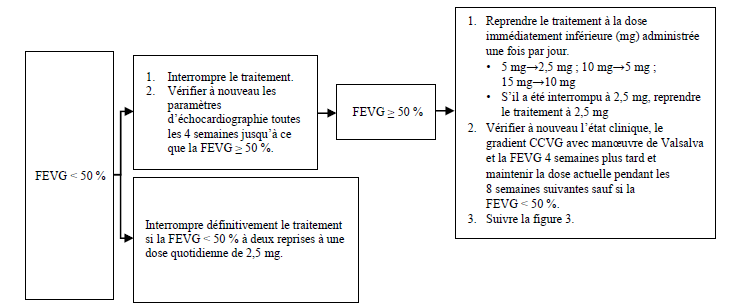
CCVG = chambre de chasse ventriculaire gauche ; FEVG = fraction d'éjection ventriculaire gauche
Modification de la posologie avec des médicaments concomitants
Suivre les étapes indiquées dans le tableau 1 pour le traitement concomitant par inhibiteurs ou inducteurs du CYP2C19 ou du CYP3A4 (voir également rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Tableau 1 : Modification de la posologie du mavacamten en fonction des traitements concomitants
|
Traitement concomitant |
Phénotype métaboliseur lent du CYP2C19* |
Phénotype métaboliseur intermédiaire, normal, rapide et ultrarapide du CYP2C19 |
|
Inhibiteurs |
||
|
Utilisation concomitante avec un inhibiteur puissant du CYP2C19 et un inhibiteur puissant du CYP3A4 |
Contre-indiqué (voir rubrique Contre-indications). |
Contre-indiqué (voir rubrique Contre-indications). |
|
Inhibiteur puissant du CYP2C19 |
Aucun ajustement posologique (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Si le phénotype du CYP2C19 n'a pas encore été déterminé : Aucun ajustement de la dose initiale de 2,5 mg n'est nécessaire. Réduire la dose de 5 mg à 2,5 mg, ou interrompre le traitement s'il est à 2,5 mg (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
Instaurer le mavacamten à une dose de 2,5 mg. Réduire la dose de 15 mg à 5 mg, de 10 mg à 2,5 mg et de 5 mg à 2,5 mg, ou interrompre le traitement s'il est à 2,5 mg (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
|
Inhibiteur puissant du CYP3A4 |
Contre-indiqué (voir rubrique Contre-indications). |
Aucun ajustement posologique (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
|
Inhibiteur modéré du CYP2C19 |
Aucun ajustement posologique. Si le phénotype du CYP2C19 n'a pas encore été déterminé : Aucun ajustement de la dose initiale de 2,5 mg n'est nécessaire. Réduire la dose de 5 mg à 2,5 mg ou interrompre le traitement s'il est à 2,5 mg (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
Aucun ajustement de la dose initiale de 5 mg n'est nécessaire. Réduire la dose d'un niveau de dose ou interrompre le traitement s'il est à 2,5 mg (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
|
Inhibiteur modéré ou faible du CYP3A4 |
Aucun ajustement de la dose initiale de 2,5 mg n'est nécessaire. Si le patient reçoit une dose de 5 mg de mavacamten, réduire sa dose à 2,5 mg (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
Aucun ajustement posologique (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
|
Inducteurs |
||
|
Interruption ou réduction de la dose d'un inducteur puissant du CYP2C19 et d'un inducteur puissant du CYP3A4 |
Réduire la dose de 5 mg à 2,5 mg ou interrompre le traitement s'il est à 2,5 mg (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
En cas d'interruption ou de réduction de la dose des inducteurs puissants lors du traitement par mavacamten, réduire la dose d'un niveau de dose lorsque le traitement est à 5 mg ou plus (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Aucun ajustement posologique si le traitement est à 2,5 mg. |
|
Interruption ou réduction de la dose d'un inducteur modéré ou faible du CYP3A4 |
Réduire la dose de mavacamten à 2,5 mg ou interrompre le traitement s'il est à 2,5 mg (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
Aucun ajustement posologique (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). |
* inclut les patients dont le phénotype du CYP2C19 n'a pas encore été déterminé.
Doses oubliées ou retardées
Si une dose est oubliée, elle doit être prise dès que possible et la prochaine dose programmée doit être prise à l'heure habituelle le jour suivant. Ne pas prendre deux doses le même jour.
Populations particulières
Personnes âgées
Aucun ajustement posologique de la dose standard et du calendrier de titration n'est requis pour les patients âgés de 65 ans et plus (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénaleAucun ajustement posologique de la dose standard et du calendrier de titration n'est requis pour les patients présentant une insuffisance rénale légère (débit de filtration glomérulaire estimé [DFG estimé] de 60-89 mL/min/1,73 m2) à modérée (DFG estimé de 30-59 mL/min/1,73 m2). Aucune recommandation posologique ne peut être donnée pour les patients présentant une insuffisance rénale sévère (DFG estimé < 30 mL/min/1,73 m2), le mavacamten n'ayant pas été étudié chez les patients atteints d'une insuffisance rénale sévère (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
La posologie initiale du mavacamten doit être de 2,5 mg chez tous les patients présentant une insuffisance hépatique légère (Child-Pugh classe A) ou modérée (Child-Pugh classe B), puisqu'il est probable que l'exposition au mavacamten soit augmentée (voir rubrique Propriétés pharmacocinétiques). Aucune recommandation posologique ne peut être donnée pour les patients atteints d'une insuffisance hépatique sévère (Child-Pugh classe C), le mavacamten n'ayant pas été étudié chez les patients atteints d'une insuffisance hépatique sévère (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité du mavacamten chez les enfants et les adolescents de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Le mavacamten ne doit pas être utilisé chez les enfants âgés de moins de 12 ans en raison de potentiels problèmes de sécurité.
Mode d'administration
Voie orale.
Le traitement doit être pris une fois par jour, au cours ou entre les repas, à la même heure environ chaque jour. La gélule doit être avalée en entier avec de l'eau.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Les données de surdosage observées avec le mavacamten chez l'homme sont restreintes. Le mavacamten a été administré à une dose unique allant jusqu'à 144 mg chez des patients atteints d'une CMH. Un effet indésirable grave de réaction vasovagale, hypotension et asystole de 38 secondes a été rapporté à cette dose. Chez les sujets sains, des doses allant jusqu'à 25 mg ont été administrées pendant une période maximale de 25 jours. Une diminution de la FEVG de 20 % ou plus a été enregistrée chez 3 des 8 participants traités au niveau de dose de 25 mg. Le dysfonctionnement systolique est le résultat le plus probable de surdosage du mavacamten. Si nécessaire, le traitement du surdosage par mavacamten consiste à interrompre le traitement par mavacamten et à apporter des soins de soutien afin de maintenir l'état hémodynamique (par ex. instauration d'un soutien inotrope avec des agents adrénergiques), y compris la surveillance étroite des signes vitaux et de la FEVG, et la prise en charge de l'état clinique du patient.
Chez les sujets sains à jeun pendant une nuit, l'administration de charbon actif 2 heures (environ tmax) après l'ingestion d'une dose de 15 mg de mavacamten a réduit l'absorption telle qu'exprimée par l'ASC0-72 de 20 %. L'administration de charbon actif 6 heures après la dose de mavacamten n'a pas eu d'effet sur l'absorption. Par conséquent, l'administration précoce (avant ou dès que possible après le tmax) de charbon actif peut être envisagée pour la prise en charge d'un surdosage ou d'une ingestion accidentelle de mavacamten. En présence de nourriture, le charbon actif peut rester efficace plus de 2 heures après la dose de mavacamten car le tmax est retardé (voir rubrique Propriétés pharmacocinétiques).
Classe pharmacothérapeutique : médicaments en cardiologie, autres préparations cardiaques, Code ATC : C01EB24
Mécanisme d'action
Le mavacamten est un inhibiteur sélectif, allostérique et réversible de la myosine cardiaque. Le mavacamten module le nombre de têtes de myosine qui peuvent entrer dans un état générateur d'énergie, réduisant ainsi (ou, dans le cas de la CMH, normalisant) la probabilité de formation de ponts croisés systoliques et diastoliques résiduels générateurs de force. Le mavacamten fait également évoluer la population globale de têtes de myosine dans un état super relaxé, économe en énergie, mais dans lequel elles restent mobilisables. La formation excessive de ponts croisés et le dérèglement de l'état super relaxé de la myosine sont des caractéristiques mécanistiques de la CMH, qui peuvent entraîner une hypercontractilité, une relaxation altérée, une consommation d'énergie excessive et une contrainte sur la paroi myocardique. Chez les patients atteints d'une CMH, l'inhibition de la myosine cardiaque par le mavacamten normalise la contractilité, réduit l'obstruction dynamique de la chambre de chasse du ventricule gauche (CCVG) et améliore les pressions de remplissage cardiaque.
Effets pharmacodynamiques
FEVG
Dans
l'étude EXPLORER-HCM, la FEVG moyenne (écart-type) au repos était de 74 % (6) à
l'inclusion pour les deux groupes de traitement, les diminutions de la
variation absolue moyenne de la FEVG par rapport à l'inclusion étaient de -4 %
(IC à 95 % : -5,3, -2,5) pour le groupe mavacamten et
de 0 % (IC à 95 % : -1,2, 1,0) pour le groupe placebo, pendant la période de
traitement de 30 semaines. À la semaine 38, la FEVG moyenne après une
interruption du traitement par mavacamten de 8
semaines, était similaire à la FEVG moyenne à l'inclusion pour les deux groupes
de traitement.
Obstruction CCVG
Dans
l'étude EXPLORER-HCM, les patients ont obtenu des diminutions du gradient CCVG
moyen au repos et provoqué (Valsalva) à la semaine 4, qui se sont maintenues
tout au long de la durée de l'étude de 30 semaines. À la semaine 30, les
variations moyennes des gradients CCVG au repos et avec manœuvre de Valsalva
par rapport à l'inclusion étaient de -39 (IC à 95 % : -44,0, -33,2) mmHg et -49 (IC à 95 % : -55,4, -43,0) mmHg,
respectivement, pour le groupe mavacamten et -6 (IC à
95 % : -10,5, -0,5) mmHg et -12 (IC à 95 % : -17,6,
-6,6) mmHg, respectivement, pour le groupe placebo. À
la semaine 38, après 8 semaines d'arrêt du mavacamten,
la FEVG et les gradients CCVG moyens étaient similaires aux valeurs à
l'inclusion pour les deux groupes de traitement.
Électrophysiologie cardiaque
Dans
la CMH, l'intervalle QT peut être prolongé intrinsèquement par la maladie
sous-jacente, en association avec la stimulation ventriculaire, ou en
association avec des médicaments susceptibles d'allonger l'intervalle QT,
couramment utilisés dans la population atteinte de CMH. Une analyse
exposition/réponse portant sur l'ensemble des études cliniques réalisées auprès
de patients atteints de CMH a mis en évidence un raccourcissement de
l'intervalle QTcF avec le mavacamten
dépendant de la concentration. La variation moyenne corrigée par le placebo par
rapport à l'inclusion chez les patients atteints d'une CMHo
était de -8,7 ms (limite supérieure et inférieure de l'IC à 90 % -6,7 ms et
-10,8 ms, respectivement) à la Cmax médiane à l'état
d'équilibre de 452 ng/mL.
Les patients qui présentaient les intervalles QTcF
initiaux les plus longs avaient tendance à enregistrer le plus grand
raccourcissement.
Conformément aux résultats non cliniques obtenus sur des cœurs normaux, une étude clinique menée chez des sujets sains a révélé que l'exposition prolongée au mavacamten à des niveaux supérieurs aux concentrations thérapeutiques et entraînant une dépression marquée de la fonction systolique, était associée à un allongement de l'intervalle QTc (< 20 ms). Aucune modification aiguë de l'intervalle QTc n'a été observée à des expositions comparables (ou supérieures) après des doses uniques. Les résultats obtenus sur des cœurs normaux sont attribués à une réponse adaptative aux changements mécaniques/fonctionnels cardiaques (dépression mécanique marquée du VG) se produisant en réponse à l'inhibition de la myosine dans les cœurs ayant une physiologie et une contractilité normales du VG.
Efficacité et sécurité cliniques
Étude EXPLORER-HCM
L'efficacité
du mavacamten a été évaluée dans une étude de phase
III en double aveugle, randomisée, contrôlée versus placebo, en groupes
parallèles, multicentrique et internationale, réalisée auprès de 251 patients
adultes atteints d'une CMHo de classe NYHA II et III
et présentant une FEVG ≥ 55 %, un gradient CCVG maximal ≥ 50 mmHg au repos ou avec provocation au moment du diagnostic
de la CMHo, et un gradient CCVG avec manœuvre de
Valsalva ≥ 30 mmHg à la sélection. La majorité
des patients ont reçu le traitement de fond de la CMH, pour un total de 96 %
pour le groupe mavacamten (bêta-bloquants
76 %, inhibiteurs des canaux calciques 20 %) et de 87 % pour le groupe placebo
(bêta- bloquants 74 %, inhibiteurs des canaux calciques 13 %).
Les
patients ont été randomisés selon un rapport 1:1 pour recevoir une dose
initiale de mavacamten de 5 mg (123 patients) ou un
placebo correspondant (128 patients) une fois par jour pendant 30 semaines.
La
dose était ajustée régulièrement afin d'optimiser la réponse des patients
(diminution du gradient CCVG évalué avec manœuvre de Valsalva) et de maintenir
la FEVG ≥ 50 % ; elle était également guidée par les concentrations
plasmatiques du mavacamten. Pour les posologies
allant de 2,5 mg à 15 mg, un total de 60 patients ont reçu 5 mg et 40 patients
ont reçu 10 mg. Pendant l'étude, 3 des 7 patients sous mavacamten
ont enregistré une FEVG < 50 % avant la visite à la semaine 30 et ont
interrompu temporairement leur traitement ; 2 patients ont redémarré le
traitement à la même dose et 1 patient a vu sa dose réduite de 10 mg à 5 mg.
L'attribution des traitements était stratifiée par la classe NYHA (II ou III) initiale, traitement en cours par des bêta-bloquants (oui ou non) et type d'ergomètre (tapis roulant ou bicyclette ergométrique) utilisé pour l'évaluation de la consommation maximale d'oxygène (VO2 max). Les patients sous bithérapie de bêta-bloquants et d'inhibiteurs des canaux calciques ou disopyramide ou ranolazine, étaient exclus. Les patients atteints d'un trouble infiltrant ou d'une maladie de surcharge provoquant une hypertrophie cardiaque qui ressemblerait à la CMHo, comme la maladie de Fabry, l'amylose ou le syndrome de Noonan avec hypertrophie du VG, étaient également exclus.
Les caractéristiques démographiques et pathologiques initiales étaient équilibrées entre le groupe mavacamten et le groupe placebo. L'âge moyen était de 59 ans, 54 % (mavacamten) contre 65 % (placebo) étaient de sexe masculin, l'indice de masse corporelle (IMC) moyen était de 30 kg/m2, la fréquence cardiaque moyenne de 63 bpm, la pression artérielle moyenne de 128/76 mmHg, et 90 % étaient d'origine caucasienne. À l'inclusion, environ 73 % des sujets randomisés étaient en classe NYHA II et 27 % en classe NYHA III. Le gradient moyen avec manœuvre de Valsalva était de 73 mmHg. Huit pour cent avaient déjà subi une thérapie de réduction septale, 75 % étaient sous bêta- bloquants, 17 % étaient sous inhibiteurs des canaux calciques, 14 % avaient des antécédents de fibrillation atriale, et 23 % étaient porteurs d'un défibrillateur-cardioverteur implantable (23 %). Dans l'étude EXPLORER-HCM, 85 patients étaient âgés de 65 ans ou plus et 45 patients ont été traités par mavacamten.
Le critère d'évaluation principal comprenait la variation de la capacité physique à la semaine 30, mesurée par la VO2 max, et les symptômes, mesurés par la classification fonctionnelle NYHA (définis comme une amélioration de la VO2 max de ≥ 1,5 mL/kg/min et une amélioration de la classe NYHA d'un moins 1 OU une amélioration de la VO2 max de ≥ 3,0 mL/kg/min et sans aggravation de la classe NYHA).
Une proportion plus importante de patients traités par mavacamten a atteint les critères d'évaluation principaux et secondaires à la semaine 30 par rapport au placebo (voir tableau 4).
Tableau 4 : Analyse des critères composites principaux et
secondaires de
l'étude EXPLORER-HCM
|
Mavacamten N = 123 |
Placebo
N = 128 |
|
|
Patients
atteignant le critère d'évaluation principal à la semaine 30, n (%) |
45 (37 %) | 22 (17 %) |
| Différence entre les traitements (IC à 95 %) | 19,4 (8,67, 30,13) | |
| Valeur de p | 0,0005 | |
|
Variation
du gradient CCVG maximal post-exercice entre l'inclusion et la semaine 30, mmHg |
N = 123 | N = 128 |
| Moyenne (écart-type) | -47 (40) | -10 (30) |
| Différence entre les traitements* (IC à 95 %) | -35 (-43, -28) | |
| Valeur de p | < 0,0001 | |
|
Variation
de la VO2 max entre l'inclusion et la semaine 30, mL/kg/min |
N = 123 | N = 128 |
| Moyenne (écart-type) | 1,4 (3) | -0,05 (3) |
| Différence entre les traitements* (IC à 95 %) | 1,4 (0,6, 2) | |
| Valeur de p | < 0,0006 | |
| Patients avec amélioration de la classe NYHA ≥ 1 à la semaine 30 | N = 123 | N = 128 |
| N, (%) | 80 (65 %) | 40 (31 %) |
| Différence entre les traitements (IC à 95 %) | 34 (22, 45) | |
| Valeur de p | < 0,0001 | |
|
Variation
du score CSS du questionnaire KCCQ-23 entre l'inclusion et la semaine 30† |
N = 92 | N = 88 |
| Moyenne (écart-type) | 14 (14) | 4 (14) |
| Différence entre les traitements* (IC à 95 %) | 9 (5, 13) | |
| Valeur de p | < 0,0001 | |
| Inclusion | N = 99 | N = 97 |
| Moyenne (écart-type) | 71 (16) | 71 (19) |
|
Variation
du score de domaine de l'essoufflement du questionnaire HCMSQ entre l'inclusion et la semaine 30‡ |
N = 85 | N = 86 |
| Moyenne (écart-type) | -2,8 (2,7) | -0,9 (2,4) |
| Différence entre les traitements* (IC à 95 %) | -1,8 (-2,4, -1,2) | |
| Valeur de p | < 0,0001 | |
| Inclusion | N = 108 | N = 109 |
| Moyenne (écart-type) | 4,9 (2,5) | 4,5 (3,2) |
*
Différence des moindres carrés
† Score CSS du questionnaire KCCQ-23 = score clinique global du Kansas City Cardiomyopathy
Questionnaire-23. Le score CSS du questionnaire KCCQ-23 est dérivé du score total des symptômes (TSS) et du score des restrictions physiques (RP) du questionnaire KCCQ-23. Le score CSS varie de 0 à 100, les scores les plus élevés représentant un meilleur état de santé. Un effet significatif du traitement sur le score CSS du questionnaire KCCQ-23 favorisant le mavacamten a d'abord été observé à la semaine 6 et s'est stabilisé jusqu'à la semaine 30.
‡ HCMSQ SoB = Hypertrophic Cardiomyopathy Symptom Questionnaire Shortness of Breath. Le score de domaine de l'essoufflement du questionnaire HCMSQ SoB mesure la fréquence et la sévérité de l'essoufflement. Le score de domaine de l'essoufflement du questionnaire HCMSQ SoB varie de 0 à 18, les scores les plus faibles représentant un essoufflement moins important. Un effet significatif du traitement sur l'essoufflement du questionnaire HCMSQ SoB favorisant le mavacamten a d'abord été observé à la semaine 4 et s'est stabilisé jusqu'à la semaine 30.
† Score CSS du questionnaire KCCQ-23 = score clinique global du Kansas City Cardiomyopathy
Questionnaire-23. Le score CSS du questionnaire KCCQ-23 est dérivé du score total des symptômes (TSS) et du score des restrictions physiques (RP) du questionnaire KCCQ-23. Le score CSS varie de 0 à 100, les scores les plus élevés représentant un meilleur état de santé. Un effet significatif du traitement sur le score CSS du questionnaire KCCQ-23 favorisant le mavacamten a d'abord été observé à la semaine 6 et s'est stabilisé jusqu'à la semaine 30.
‡ HCMSQ SoB = Hypertrophic Cardiomyopathy Symptom Questionnaire Shortness of Breath. Le score de domaine de l'essoufflement du questionnaire HCMSQ SoB mesure la fréquence et la sévérité de l'essoufflement. Le score de domaine de l'essoufflement du questionnaire HCMSQ SoB varie de 0 à 18, les scores les plus faibles représentant un essoufflement moins important. Un effet significatif du traitement sur l'essoufflement du questionnaire HCMSQ SoB favorisant le mavacamten a d'abord été observé à la semaine 4 et s'est stabilisé jusqu'à la semaine 30.
Une série de caractéristiques démographiques, de caractéristiques pathologiques initiales et de médicaments concomitants pris à l'inclusion ont été examinés pour leur retentissement sur les résultats. Les résultats de l'analyse principale étaient systématiquement en faveur du mavacamten pour tous les sous-groupes analysés.
Étude VALOR-HCM
L'efficacité
du mavacamten a été évaluée dans une étude de phase
III de 16 semaines, randomisée, contrôlée versus placebo, menée en
double aveugle auprès de 112 patients atteints de CMHo
symptomatique qui étaient éligibles à la thérapie de réduction septale (TRS).
Les patients atteints de CMHo symptomatique sévère
réfractaire au traitement et de classe NYHA III/IV ou NYHA II avec syncope ou
quasi-syncope à l'effort ont été inclus dans l'étude. Les patients devaient
présenter un gradient CCVG maximal ≥ 50 mmHg au
repos ou avec provocation, et une FEVG ≥ 60 %. Les patients devaient
avoir été adressés par un professionnel de santé ou avoir fait l'objet d'un
examen approfondi dans les 12 derniers mois aux fins de la TRS et ont
activement envisagé la programmation de la procédure.
Les patients ont été randomisés selon un rapport de 1:1 pour recevoir le traitement par mavacamten ou le placebo une fois par jour. La dose était ajustée régulièrement dans l'intervalle de doses de 2,5 mg à 15 mg afin d'optimiser la réponse du patient.
Les caractéristiques initiales démographiques et relatives à la maladie étaient équilibrées entre le groupe mavacamten et le groupe placebo. L'âge moyen était de 60,3 ans, 51 % étaient de sexe masculin, l'IMC moyen était de 31 kg/m2, la fréquence cardiaque moyenne de 64 bpm, la pression artérielle moyenne de 131/74 mmHg, et 89 % étaient d'origine caucasienne. À l'inclusion, environ 7 % des sujets randomisés étaient de classe NYHA II et 92 % étaient de classe NYHA III. 46 % étaient sous monothérapie par bêta-bloquants, 15 % étaient sous monothérapie par inhibiteurs des canaux calciques, 33 % étaient sous bithérapie de bêta-bloquants et d'inhibiteurs des canaux calciques, et 20 % étaient sous disopyramide seul ou en association avec d'autres traitements. Dans l'étude VALOR-HCM, 45 patients étaient âgés de 65 ans ou plus et 24 patients ont été traités par mavacamten.
Le
mavacamten s'est montré supérieur au placebo dans la
réponse au critère composite principal à la semaine 16 (voir tableau 5). Le
critère d'évaluation principal était un critère composite de
- la proportion de patients décidant de procéder à une TRS à la semaine 16 ou avant, ou
- la proportion de patients demeurant éligibles à la TRS (gradient CCVG ≥ 50 mmHg et classe NYHA III/IV ou classe NYHA II avec syncope ou quasi-syncope à l'effort) à la semaine 16.
Les effets du traitement par mavacamten sur l'obstruction de la CCVG, la capacité fonctionnelle, l'état de santé et les biomarqueurs cardiaques ont été évalués entre l'inclusion et la semaine 16 à partir de la variation post-exercice des paramètres suivants : gradient CCVG, proportion de patients présentant une amélioration de la classe NYHA, score CSS du questionnaire KCCQ-23, NT-proBNP et troponine cardiaque I. Dans l'étude VALOR-HCM, les tests hiérarchiques des critères d'efficacité secondaires ont montré une amélioration significative dans le groupe mavacamten par rapport au groupe placebo (voir tableau 5).
Tableau 5 : Analyse des critères composites principaux et
secondaires de
l'étude VALOR-HCM
|
Mavacamten N = 56 |
Placebo
N = 56 |
|
|
Patients
atteignant le critère composite principal à la semaine 16, n (%) |
10 (17,9) | 43 (76,8) |
| Différence entre les traitements (IC à 95 %) | 58,9 (44,0 ; 73,9) | |
| Valeur de p | < 0,0001 | |
| Décision du patient de procéder à une TRS | 2 (3,6) | 2 (3,6) |
| Éligibilité à la TRS selon les critères des recommandations | 8 (14,3) | 39 (69,6) |
|
Statut
de la TRS impossible à évaluer (imputé comme répondant au critère d'évaluation principal) |
0 (0,0) | 2 (3,6) |
|
Variation
du gradient CCVG maximal post-exercice entre l'inclusion et la semaine 16, (mmHg) |
N = 55 | N = 53 |
| Moyenne (écart-type) | -39,1 (36,5) | -1,8 (28,8) |
| Différence entre les traitements* (IC à 95 %) | -37,2 (-48,1 ; -26,2) | |
| Valeur de p | < 0,0001 | |
|
Patients
présentant une amélioration de la classe NYHA ≥ 1 à la semaine 16 |
N = 55 | N = 53 |
| N (%) | 35 (62,5 %) | 12 (21,4 %) |
| Différence entre les traitements (IC à 95 %) | 41,1 (24,5 %, 57,7 %) | |
| Valeur de p | < 0,0001 | |
|
Variation
du score CSS du questionnaire KCCQ-23 entre l'inclusion et la semaine 16† |
N = 55 | N = 53 |
| Moyenne (écart-type) | 10,4 (16,1) | 1,8 (12,0) |
| Différence entre les traitements* (IC à 95 %) | 9,5 (4,9 ; 14,0) | |
| Valeur de p | < 0,0001 | |
| Inclusion | N = 56 | N = 56 |
| Moyenne (écart-type) | 69,5 (16,3) | 65,6 (19,9) |
| Variation du NT-proBNP entre l'inclusion et la semaine 16 | N = 55 | N = 53 |
| Ratio des moyennes géométriques en ng/L | 0,35 | 1,13 |
|
Ratio
des moyennes géométriques pour le mavacamten/placebo
(IC à 95 %) |
0,33 (0,27 ; 0,42) | |
| Valeur de p | < 0,0001 | |
|
Variation
de la troponine cardiaque I entre l'inclusion et la semaine 16 |
N = 55 | N = 53 |
| Ratio des moyennes géométriques en ng/L | 0,50 | 1,03 |
|
Ratio
des moyennes géométriques pour le mavacamten/placebo
(IC à 95 %) |
0,53 (0,41 ; 0,70) | |
| Valeur de p | < 0,0001 | |
*
Différence des moindres carrés.
† Score CSS du questionnaire KCCQ-23 = score clinique global du Kansas City Cardiomyopathy
Questionnaire-23. Le score CSS du questionnaire KCCQ-23 est dérivé du score total des symptômes (TSS) et du score des restrictions physiques (RP) du questionnaire KCCQ-23. Le score CSS varie de 0 à 100, les scores les plus élevés représentant un meilleur état de santé.
† Score CSS du questionnaire KCCQ-23 = score clinique global du Kansas City Cardiomyopathy
Questionnaire-23. Le score CSS du questionnaire KCCQ-23 est dérivé du score total des symptômes (TSS) et du score des restrictions physiques (RP) du questionnaire KCCQ-23. Le score CSS varie de 0 à 100, les scores les plus élevés représentant un meilleur état de santé.
Dans
l'étude VALOR-HCM, le critère d'évaluation secondaire du NT-proBNP
à la semaine 16 (voir tableau 5) a montré une diminution durable, à partir de
l'inclusion après le traitement par mavacamten par
rapport au placebo, similaire à celle observée dans l'étude EXPLORER-HCM à la
semaine 30.
L'analyse
exploratoire de l'indice de masse ventriculaire gauche (IMVG) et de l'indice de
volume atrial gauche (IVAG) a montré des réductions chez les patients traités
par mavacamten par rapport au placebo dans les études
EXPLORER-HCM et VALOR-HCM.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec CAMZYOS dans un ou plusieurs sous-groupes de la population pédiatrique pour le traitement de la CMH (voir rubrique Posologie et mode d'administration pour les informations concernant l'utilisation pédiatrique).
Absorption
Le mavacamten est facilement absorbé, avec un tmax médian de 1 heure (de 0,5 à 3 heures) après administration orale et une biodisponibilité orale estimée à environ 85 % dans la plage de doses cliniques. L'augmentation de l'exposition au mavacamten est généralement proportionnelle à la dose, après des doses une fois par jour de mavacamten (2 mg à 48 mg).
Après une dose unique de 15 mg de mavacamten, la Cmax et l'ASCinf sont 47 % et 241 % plus élevées, respectivement, chez les métaboliseurs lents du CYP2C19 par rapport aux métaboliseurs normaux. La demi-vie moyenne est prolongée chez les métaboliseurs lents du CYP2C19 par rapport aux métaboliseurs normaux (23 jours versus 6 à 9 jours, respectivement).
La variabilité PK interindividuelle est modérée, avec un coefficient de variation pour l'exposition d'environ 30-50 % pour la Cmax et l'ASC.
Un repas riche en graisses et en calories a retardé l'absorption, avec un tmax médian de 4 heures (de 0,5 à 8 heures) lorsque les patients étaient alimentés versus un tmax médian de 1 h à jeun. L'administration au cours des repas a entraîné une diminution de 12 % de l'ASC0-inf, mais cette diminution n'est pas considérée comme cliniquement significative. Le mavacamten peut être administré au cours ou en dehors des repas.
La posologie du mavacamten étant ajustée en fonction de la réponse clinique (voir rubrique Posologie et mode d'administration), les expositions à l'état d'équilibre simulées sont résumées sur la base de la posologie individualisée en fonction du phénotype (voir tableau 6).
Tableau
6 : Concentration moyenne simulée à l'état d'équilibre selon la dose et
le phénotype
du CYP2C19 chez les patients dont la dose a été ajustée
pour obtenir l'effet
souhaité sur la base de la CCVG avec manœuvre de
Valsalva et de la FEVG
| Dose | Concentration médiane (ng/mL) | ||||
| Métaboliseurs lents | Métaboliseurs intermédiaires | Métaboliseurs normaux | Métaboliseurs rapides | Métaboliseurs ultrarapides | |
| 2,5 mg | 451,9 | 274,0 | 204,9 | 211,3 | 188,3 |
| 5 mg | 664,9 | 397,8 | 295,4 | 311,5 | 300,5 |
Distribution
La liaison aux protéines plasmatiques du mavacamten est de 97-98 % dans les études cliniques. Le rapport de concentration sang/plasma est de 0,79. Le volume de distribution apparent (Vd/F) allait de 114 L à 206 L. Aucune étude spécifique pour évaluer la distribution du mavacamten n'a été menée chez l'homme, mais les données disponibles sont cohérentes avec un volume de distribution élevé.
Sur la base de 10 sujets de sexe masculin ayant été traités pendant une durée maximale de 28 jours, la quantité de mavacamten distribuée dans le sperme a été considérée comme faible.
Biotransformation
Le mavacamten est largement métabolisé, principalement par le CYP2C19 (74 %), le CYP3A4 (18 %) et le CYP2C9 (7,6 %) d'après le phénotypage in vitro. Le métabolisme devrait faire intervenir les trois voies, et principalement celle du CYP2C19 chez les métaboliseurs intermédiaires, normaux, rapides et ultrarapides du CYP2C19. Trois métabolites ont été détectés dans le plasma humain. L'exposition du métabolite le plus abondant, le MYK-1078, dans le plasma humain, était inférieure à 4 % de l'exposition du mavacamten, et les deux autres métabolites enregistraient des expositions inférieures à 3 % de l'exposition du mavacamten, indiquant que ceux-ci auraient une incidence minime, voire nulle, sur l'activité globale du mavacamten. Chez les métaboliseurs lents du CYP2C19, le mavacamten est principalement métabolisé par le CYP3A4. Aucune donnée sur le profil métabolique des métaboliseurs lents du CYP2C19 n'est disponible.
Effet du mavacamten sur d'autres enzymes CYP
Selon
les données pré-cliniques, pour une dose allant jusqu'à 5 mg chez les
métaboliseurs lents du CYP2C19 et jusqu'à 15 mg chez les métaboliseurs
intermédiaires à ultrarapides du CYP2C19, le mavacamten n'est pas un
inhibiteur du CYP 1A2, 2B6, 2C8, 2D6, 2C9, 2C19 ou 3A4 à des
concentrations cliniquement pertinentes.
Effet du mavacamten sur les transporteurs
Les données in vitro indiquent
que, pour une dose allant jusqu'à 5 mg chez les métaboliseurs lents du
CYP2C19 et jusqu'à 15 mg chez les métaboliseurs intermédiaires à
ultrarapides du CYP2C19, le mavacamten n'est pas un inhibiteur des
principaux transporteurs d'efflux (P-gp, BCRP, BSEP, MATE1 ou MATE2-K)
ou des principaux transporteurs d'absorption (polypeptides
transporteurs d'anions organiques [OATP], transporteurs de cations
organiques [OCT] ou transporteurs d'anions organiques [OAT]) à des
concentrations thérapeutiques.
Élimination
Le
mavacamten est éliminé du plasma principalement par métabolisation par
les enzymes du cytochrome P450. Sa demi-vie terminale est de 6 à 9
jours chez les métaboliseurs normaux du CYP2C19 et de 23 jours chez les
métaboliseurs lents du CYP2C19.
Sa demi-vie est estimée à 6 jours chez les métaboliseurs ultrarapides
du CYP2C19, 8 jours chez les métaboliseurs rapides du CYP2C19 et 10
jours chez les métaboliseurs intermédiaires du CYP2C19.
L'accumulation de médicament se produit dans un rapport d'accumulation d'environ 2 fois pour la Cmax et d'environ 7 fois pour l'ASC chez les métaboliseurs normaux du CYP2C19. L'accumulation dépend du statut de métaboliseur du CYP2C19, avec l'accumulation la plus importante observée chez les métaboliseurs lents du CYP2C19. À l'état d'équilibre, le rapport entre la concentration plasmatique maximale et la concentration plasmatique minimale avec une administration une fois par jour est d'environ 1,5.
Après une dose unique de 25 mg de mavacamten marqué au 14C chez des métaboliseurs normaux du CYP2C19, 7 % et 85 % de la radioactivité totale ont été retrouvés respectivement dans les selles et l'urine des métaboliseurs normaux du CYP2C19. La substance active inchangée représentait environ 1 % et 3 % de la dose administrée dans les selles et l'urine, respectivement.
Phénotype du CYP2C19
Le
CYP2C19 polymorphe constitue la principale enzyme impliquée dans le
métabolisme du mavacamten. Un individu porteur de deux allèles avec une
fonction normale est un métaboliseur normal du CYP2C19 (par ex. *1/*1).
Un individu porteur de deux allèles défectueux est un métaboliseur lent
du CYP2C19 (par ex. *2/*2, *2/*3, *3/*3).
L'incidence du phénotype métaboliseur lent du CYP2C19 varie d'environ 2
% dans les populations caucasiennes à 18 % dans les populations
asiatiques.
Linéarité/non-linéarité
L'exposition au mavacamten a augmenté à peu près proportionnellement à la dose entre 2 mg et 48 mg et devrait entraîner une augmentation de l'exposition proportionnelle à la dose dans l'intervalle thérapeutique de 2,5 mg à 5 mg chez les métaboliseurs lents du CYP2C19 et de 2,5 mg à 15 mg chez les métaboliseurs intermédiaires à ultrarapides du CYP2C19.
Populations particulières
Aucune différence cliniquement significative dans la PK du mavacamten n'a été observée en utilisant la modélisation PK de la population basée sur l'âge, le sexe, la race ou l'origine ethnique.
Insuffisance hépatique
Une
étude PK de dose unique a été menée chez des patients atteints d'une
insuffisance hépatique légère (Child-Pugh classe A) ou modérée
(Child-Pugh classe B), ainsi qu'un groupe contrôle présentant une
fonction hépatique normale. Les expositions (ASC) au mavacamten ont
augmenté de 3,2 fois et 1,8 fois chez les patients atteints d'une
insuffisance légère et modérée, respectivement, par rapport aux
patients présentant une fonction hépatique normale. La fonction
hépatique n'a eu aucun effet sur la Cmax,
ce qui est cohérent avec l'absence de modification du taux d'absorption
et/ou du volume de distribution. La quantité de mavacamten excrétée
dans l'urine dans les 3 groupes étudiés était de 3 %. Aucune étude PK
dédiée n'a été menée chez les patients atteints d'une insuffisance
hépatique sévère (Child-Pugh classe C).
Insuffisance rénale
Environ
3 % d'une dose de mavacamten est excrétée dans l'urine en tant que
substance parente. Une analyse PK de population, qui comprenait un DFG
estimé de seulement 29,5 mL/min/1,73 m2,
n'a démontré aucune corrélation entre la fonction rénale et
l'exposition. Aucune étude PK dédiée n'a été menée chez les patients
atteints d'une insuffisance rénale sévère (DFG estimé < 30
mL/min/1,73 m2).
Le mavacamten a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. L'utilisation du mavacamten peut occasionner des étourdissements. Les patients doivent être avertis de ne pas conduire des véhicules ni utiliser des machines s'ils ressentent des étourdissements.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie après administrations répétées, génotoxicité et cancérogénèse, n'ont pas révélé de risque particulier pour l'homme. Les résultats toxicologiques étaient généralement liés à des effets indésirables sur la fonction cardiaque et étaient cohérents avec les effets pharmacologiques primaires exagérés observés chez les animaux sains. Ces effets sont survenus à des expositions cliniquement pertinentes.
Toxicité pour la reproduction et fertilité
Dans les études de toxicité pour la reproduction, il n'y avait aucune preuve d'effets du mavacamten sur l'accouplement et la fertilité chez les rats mâles ou femelles ni sur la viabilité et la fertilité de la progéniture des mères quelle que soit la dose testée. Cependant, les expositions plasmatiques (ASC) du mavacamten aux doses les plus élevées testées étaient inférieures à celles chez l'homme à la dose maximale recommandée chez l'homme (DMRH).
Développement embryo-fœtal et postnatal
Le mavacamten a eu des effets défavorables sur le développement embryo-fœtal chez le rat et le lapin. Lorsque le mavacamten a été administré par voie orale à des rates gravides pendant la période d'organogenèse, une diminution du poids moyen du fœtus et des augmentations des pertes post- implantatoires et des malformations fœtales (viscérales et squelettiques), ont été observées à des expositions cliniquement pertinentes. Les malformations viscérales impliquaient une malformation cardiaque chez les fœtus, notamment un situs inversus total, tandis que les malformations squelettiques se manifestaient principalement par une incidence accrue de sternèbres fusionnées.
Lorsque le mavacamten a été administré par voie orale à des lapines gravides pendant la période d'organogenèse, des malformations viscérales et squelettiques ont été observées, à savoir des malformations des gros vaisseaux (dilatation du tronc pulmonaire et/ou de la crosse aortique), une fente palatine et une incidence plus élevée de sternèbres fusionnées. Les niveaux d'exposition plasmatique maternelle (ASC) à la dose sans effet sur le développement embryo-fœtal chez les deux espèces étaient inférieurs à ceux observés chez l'homme à la DMRH.
Dans une étude sur le développement prénatal et postnatal, l'administration du mavacamten à des rates gravides entre le jour de gestation 6 et le jour 20 de lactation/post-partum n'a pas entraîné d'effets indésirables chez les mères ou leur progéniture exposées quotidiennement avant la naissance (in utero) et au cours de la lactation. L'exposition maternelle était inférieure à la DMRH. Aucune information n'est disponible sur l'excrétion du mavacamten dans le lait animal.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste
I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription initiale hospitalière.
Prescription initiale réservée à certains spécialistes.
Renouvellement de la prescription réservé aux spécialistes en CARDIOLOGIE.
Gélule
Tête
opaque de couleur violet clair avec la mention « 2.5 mg » imprimée en
noir et corps opaque de couleur blanche avec la mention « Mava »
imprimée en noir, toutes deux dans le sens radial. Gélule d'environ
18,0 mm de longueur.
Polychlorure de vinyle (PVC) / Polychlore-trifluoroéthylène (PCTFE) / Plaquette thermoformée en aluminium contenant 14 gélules.
Boîte de 28 gélules.
Chaque gélule contient 2,5 mg de mavacamten.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule
Silice colloïdale hydratée
Mannitol (E421)
Hypromellose (E464)
Croscarmellose de sodium (E468)
Stéarate de magnésium
Enveloppe de la gélule
Gélatine
Dioxyde de titane (E171)
Oxyde de fer noir (E172)
Oxyde de fer rouge (E172)
Encre d'imprimerie
Oxyde de fer noir (E172)
Shellac (E904)
Propylène glycol (E1520)
Solution d'ammoniaque concentrée (E527)
Hydroxyde de potassium (E525)